Qin Xiang Ng, Alex Yu Sen Soh, Wayren Loke, Donovan Yutong Lim, and Wee-Song Yeo
Published online 2018 Sep 21
Abstract
Irritable bowel syndrome (IBS) is a complex, functional gastrointestinal disorder characterized by chronic abdominal pain or discomfort and altered bowel habits. Despite the global prevalence and disease burden of IBS, its underlying pathophysiology remains unclear. Inflammation may play a pathogenic role in IBS. Studies have highlighted the persistence of mucosal inflammation at the microscopic and molecular level in IBS, with increased recruitment of enteroendocrine cells. Substantial overlaps between IBS and inflammatory bowel disease have also been reported. This review thus aimed to discuss the body of evidence pertaining to the presence of mucosal inflammation in IBS, its putative role in the disease process of IBS, and its clinical relevance. Increased mast cell density and activity in the gut may correlate with symptoms of visceral hypersensitivity. As evidenced by patients who develop postinfectious IBS, infective gastroenteritis could cause systemic inflammation and altered microbiome diversity, which in turn perpetuates a cycle of chronic, low-grade, subclinical inflammation. Apart from mucosal inflammation, neuroinflammation is probably involved in the pathophysiology of IBS via the “gut–brain” axis, resulting in altered neuroendocrine pathways and glucocorticoid receptor genes. This gives rise to an overall proinflammatory phenotype and dysregulated hypothalamic–pituitary–adrenal axis and serotonergic (5-HT) functioning, which could, at least in part, account for the symptoms of IBS. Although a definite and reproducible pattern of immune response has yet to be recognized, further research into anti-inflammatories may be of clinical value.
Introduction
Irritable bowel syndrome (IBS) is a prevalent functional gastrointestinal disorder characterized by chronic abdominal pain or discomfort and altered bowel habits.1 The Rome IV criteria is frequently used for the clinical diagnosis of IBS.1 The criteria requires recurrent abdominal pain, on average, at least 1 d/wk in the last 3 months, associated with two or more of the following: 1) related to defecation; 2) associated with a change in frequency of stool; and 3) associated with a change in appearance of stools. IBS affects some 12% of the global population and has a significant disease burden in terms of increased absenteeism from school or work and reduced health-related quality of life.2,3
Despite its prevalence and often chronic, relapsing nature, the underlying pathophysiology of IBS remains incompletely understood.4 The etiology is likely multifactorial, involving dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis, neuroendocrine alterations, and visceral hypersensitivity, which results in the hallmark symptoms of abdominal pain and disordered gut motility.5 Chronic, low-grade, subclinical inflammation has been implicated in the disease process and is thought to perpetuate the symptoms of IBS.6 Previously, in most patients with IBS, routine histologic examinations did not reveal significant colonic mucosal abnormalities; however, with modern sequencing techniques, immunohistochemical assays, and ultrastructural analyses, subtle microscopic and molecular alterations have been reported.7
This review aimed to discuss the body of evidence pertaining to the presence of mucosal inflammation in IBS, its putative role in the disease process of IBS, and its clinical relevance. Evidence based on mucosal immunohistochemical examinations, patients with postinfectious IBS, human microbiota analyses, and neuroendocrine functioning would be examined and discussed. A better understanding of the role of inflammation in IBS would help advance current therapeutics and improve clinical outcomes of this difficult-to-treat condition.
Intestinal mucosa and the enteric immune system
The intestinal mucosa is part of an intricate enteric immune system and is endowed with a large variety of immune cells.8 Exposure to food, bacteria, parasites, and viruses may contribute to sensitization of the enteric immune system and activation of the inflammatory cascade. Compared to healthy controls, patients with prototypical IBS symptoms have been reported to have increased lamina propria immune cells in the colonic mucosa9 and significantly reduced levels of oleoylethanolamide, an endogenous PPAR-α agonist and fatty acid amide with anti-inflammatory properties.10 These are suggestive of chronic, low-grade inflammation at the microscopic level. Increased infiltration of mucosal mast cells have also been reported in the cecum of patients with IBS, compared to healthy controls.11 As mast cells mediate numerous inflammatory responses in the human body via activation and degranulation, it is thought that the observed increased volume density of mast cells may be the result of a persistent inflammatory response in patients with IBS. A randomized, controlled trial has found promising beneficial effects of the mast cell stabilizer ketotifen, providing further evidence for the role of mast cells and immune activation in IBS.12
Mucosal T- and B-type lymphocytes also constitute part of the gastrointestinal adaptive immune system. Colonic biopsies taken from patients with IBS have found increased density13 and activation14 of T-cell, again consistent with the hypothesis of underlying low-grade immune activation in the disease process of IBS. Increased immune activation has also been demonstrated in duodenal biopsies.15 Blood samples taken from patients with IBS also showed an activated phenotype, with an increased frequency of CD4+ and CD8+ T-cells expressing the gut homing integrin β7.16 These findings are suggestive of increased gut immune activation in IBS.
Postinfectious IBS and altered microbiome
Studies on postinfectious IBS have provided etiological insights into the pathogenesis of IBS. It is well documented that following infective gastroenteritis, more than 10% of affected individuals go on to develop postinfectious IBS.17 The risk of postinfectious IBS appears greater with bacterial gastroenteritis18 compared to viral gastroenteritis.19 Upregulated expression of IL-1β mRNA has been reported in rectal biopsies taken 3 months postinfection in patients with postinfectious IBS.20 This was not observed in patients who did not develop postinfectious IBS. IL-1β is a proinflammatory cytokine which drives cellular inflammation.
Serial rectal biopsies of patients who develop postinfectious IBS also revealed persistently increased T-cells and other enteroendocrine cells, compared to individuals whose symptoms eventually abate.21 Patients who developed postinfectious IBS also had significantly higher CD3+, CD4+, and CD8+ T-cell counts in the gastrointestinal mucosa.21 These cells are involved in the gut’s adaptive immunity response. Taken together, these findings support an inflammation–immunological etiopathogenesis of IBS.
Another consequence of infective gastroenteritis is the disruption of normal gut flora, although the gut microbiota during parasitic infection remains unstudied.17 Through the use of 16S rRNA sequencing, studies have found reduced microbial diversity in patients who develop postinfectious IBS.22 Though the gut microbiota is known to have interindividual variations, relative increase in Bacteroides and Prevotella bacteria have been frequently reported in patients with IBS compared to healthy controls.23 The microbiome is thought to modulate inflammation and act either directly or indirectly through microbial metabolites. Microbial dysbiosis has been shown to promote inflammation23 and impair normal lymphocyte function,24 ultimately perpetuating chronic, low-grade inflammation. This is supported by clinical evidence that probiotic supplementation can relieve IBS symptoms25 and have sustained effects on gut microbiota.26 Further research into methods of restoring normal gut flora, eg, via the eubiotic effects of rifaximin,27 should be encouraged. There are also existing gaps in knowledge regarding the interaction between the microbiome and the host in vivo – and the pathway of its metabolites – and how their metabolites influence the microenvironment. Metabolomic profiling28 might help answer these questions.
Overlaps between IBS and IBD
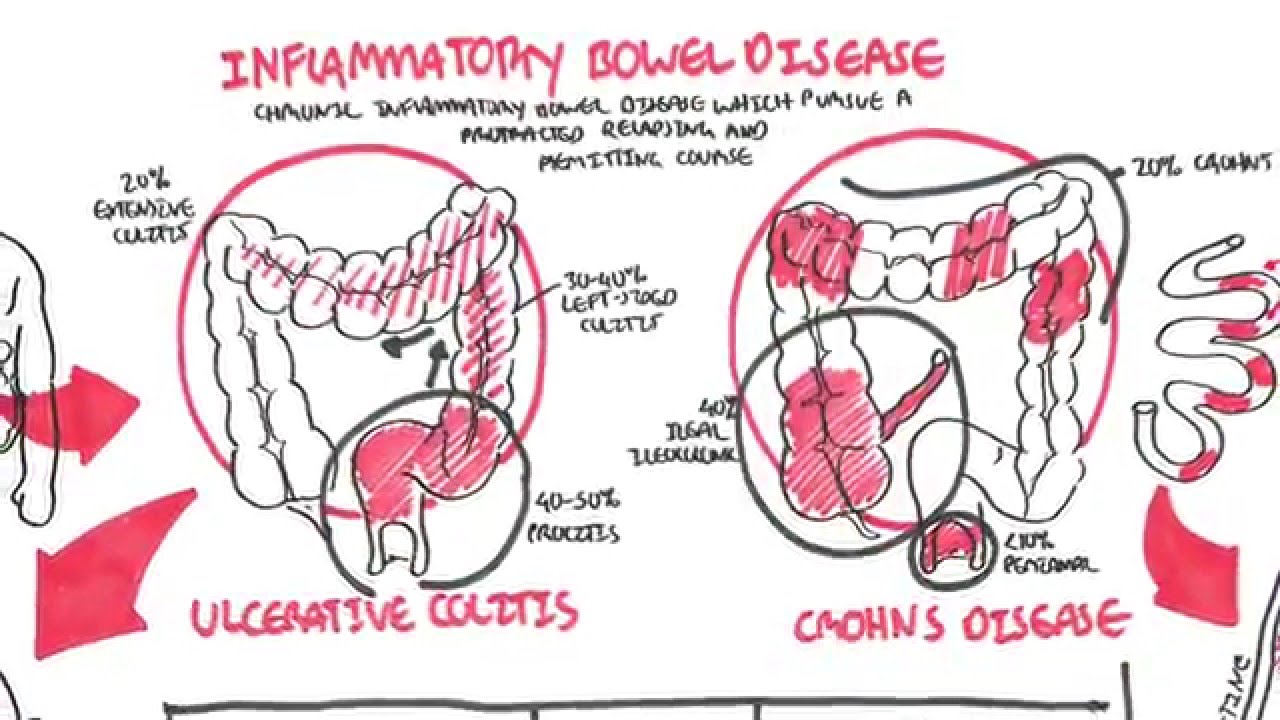
Inflammatory bowel disease (IBD) is a gastrointestinal condition characterized by chronic inflammation of the gut and colon. Although it is clear that the extent of inflammation in IBD is significantly greater than that of patients with IBS, a recent meta-analysis of 13 studies have reported a high prevalence of IBS symptoms in patients with IBD (up to 40%), even in those with quiescent disease and under remission29 (usually defined using a fecal calprotectin cutoff of 250 mg/g).30 The chronic mucosal inflammation in patients with IBD is thought to be responsible for the lasting alterations in intestinal permeability and gut function,31 thereby generating symptoms typical of IBS. When comparing mucosal biopsies taken from patients with IBS without IBD and those with IBD and IBS-like symptoms, altered intestinal permeability (specifically, decreased expression of the tight junction proteins ZO-1 and α-cathenin) was found to be a common characteristic.31 The presence of IBS-like symptoms even in patients deemed to have quiescent IBD supports the hypothesis that undetected, low-grade inflammation could be the cause of an excess of symptoms in this unique clinical population.
Neuroinflammation and the “Gut–Brain” axis
Psychosocial factors have been strongly implicated in the etiology of IBS.32 Studies have found that childhood abuse and posttraumatic stress disorder are associated with the development IBS in adulthood.33 Stress is thought to potentiate immune activation as it stimulates proinflammatory cytokines and NF-κB.34 Anxiety and mood disorders are also well-established risk factors for developing postinfectious IBS, conferring risk similar to a severe infective gastroenteritis episode.35 Modern research into the etiology of mood disorders have often alluded to the persistence of systemic and neuroinflammation.36
Early-life abuse and posttraumatic stress disorder have been shown to cause sensitized corticotropin-releasing factor systems and dysregulated HPA axis,37,38 thereby promoting a proinflammatory phenotype. Elevated levels of proinflammatory plasma cytokines IL-6 and IL-8 have been implicated in patients with IBS.39 As a result of increased proinflammatory cytokines, the activity of indoamine-2,3-dioxygenase (the rate-limiting enzyme in the degradation pathway of tryptophan) would be upregulated, ultimately affecting tryptophan metabolism and contributing to abnormal serotonergic (5-HT) functioning.40 A hyperresponsive HPA axis could contribute to visceral hypersensitivity,41 which is typically seen in patients with IBS. Abnormal 5-HT functioning is associated with altered gut motility and enhanced nociceptive pain sensitivity,42 both symptoms characteristic of IBS.
Functional magnetic resonance imaging studies have also found heightened visceral stimuli response among IBS patients, with increased activation of the anterior cingulate cortex, prefrontal cortex, and thalamus in response to rectal distention in most instances.43,44 These responses also appeared to be modulated by anxiety and depression.44
Conclusion
Current evidence does lend support to an inflammation–immunological etiopathogenesis of IBS. The various pathways discussed in this review are illustrated in Figure 1. Although a definite and reproducible pattern of immune response has yet to be recognized, increased mast cell density and activity in the gut may correlate with symptoms of visceral hypersensitivity. As evidenced by patients who develop postinfectious IBS, infective gastroenteritis could cause systemic inflammation and altered microbiome diversity, which in turn perpetuates a cycle of chronic, low-grade, subclinical inflammation. Apart from mucosal inflammation, neuroinflammation is probably involved in the pathophysiology of IBS via the “gut–brain” axis, resulting in altered neuroendocrine pathways and glucocorticoid receptor genes. This gives rise to an overall proinflammatory phenotype and dysregulated HPA axis and serotonergic (5-HT) functioning, which could, at least in part, account for the symptoms of IBS.