






19 August 2018
Li‐Rong Shao Jong M. Rho Carl E. Stafstrom
Summary
Conventional antiseizure medications reduce neuronal excitability through effects on ion channels or synaptic function. In recent years, it has become clear that metabolic factors also play a crucial role in the modulation of neuronal excitability. Indeed, metabolic regulation of neuronal excitability is pivotal in seizure pathogenesis and control.
The clinical effectiveness of a variety of metabolism‐based diets, especially for children with medication‐refractory epilepsy, underscores the applicability of metabolic approaches to the control of seizures and epilepsy. Such diets include the ketogenic diet, the modified Atkins diet, and the low‐glycemic index treatment (among others).
A promising avenue to alter cellular metabolism, and hence excitability, is by partial inhibition of glycolysis, which has been shown to reduce seizure susceptibility in a variety of animal models as well as in cellular systems in vitro. One such glycolytic inhibitor, 2‐deoxy‐d‐glucose (2DG), increases seizure threshold in vivo and reduces interictal and ictal epileptiform discharges in hippocampal slices.
Here, we review the role of glucose metabolism and glycolysis on neuronal excitability, with specific reference to 2DG, and discuss the potential use of 2DG and similar agents in the clinical arena for seizure management.
Key Points
- Cellular metabolism plays a key role in the modulation of neuronal excitability
- Inhibition of glycolysis (e.g., by 2‐deoxy‐D‐glucose, 2DG) reduces seizure occurrence and retards epilepsy progression in several animal seizure models, and abrogates epileptiform bursting in hippocampal slices
- 2DG is taken up preferentially by metabolically active cells, making this compound especially attractive for suppressing seizure activity
- 2DG appears to be safe, without long‐lasting cognitive, behavioral, or systemic effects
- Inhibitors of glycolysis offer a potential novel method for suppressing seizures and epilepsy
Neuronal excitability has been traditionally thought to be mediated predominantly via ion channels and synaptic transmission. However, it is becoming increasingly clear that metabolic factors also play a role in the modulation of neuronal excitability.1, 2 Several examples of beneficial metabolic treatments for epilepsy and other neurologic disorders are already in clinical use, including the high‐fat, low‐carbohydrate ketogenic diet (KD) and its variants (e.g., medium‐chain triglyceride diet, modified Atkins diet, and low‐glycemic index treatment).3-6 However, the mechanisms of action underlying these metabolic treatments are not fully understood.
Possible mechanisms include reduction in excitability by ketone bodies or fatty acids, altered neurotransmitter synthesis or action, improved mitochondrial function, or a variety of other factors. The observation that carbohydrate ingestion by children receiving the KD resulted in loss of seizure control7 led to the hypothesis that carbohydrate restriction could protect against seizure occurrence.8
In addition to limiting carbohydrate intake, restricting total calorie intake also suppresses seizures and affords neuroprotection.8-11 In fact, the original idea behind the KD was to mimic the physiologic effects of fasting, and some data support intermittent fasting for seizure control,12 but this strategy is not a pragmatic long‐term treatment option.
Glucose Regulation of Neuronal Excitability
The brain is a highly energy‐dependent organ, consuming at least 20% of the body's total caloric needs.13 Much of this energy demand stems from the physiologic requirements to maintain cellular ion gradients, transmembrane pumps, neurotransmitter synthesis, and other critical homeostatic processes. In this context, glucose is an obligate energy source for the brain.
All highly evolved brain functions, such as learning, cognition, and memory, are closely linked to brain glucose availability, and deficits in glucose supply and utilization have been demonstrated in a multitude of neurologic disorders, including epilepsy.14
In epilepsy, the underlying mechanisms responsible for the genesis of spontaneous recurrent seizures arise from neuronal hyperexcitability and synchrony, processes that markedly increase energy use. Fundamental to both normal and abnormal brain metabolism are astrocytes, which control both neurovascular and neurometabolic coupling by providing fuel (primarily the glucose metabolite lactate) to neurons via the lactate shuttle.13, 15, 16 In addition, cerebrovascular integrity and regulation are intimately involved with neurometabolic fluctuations associated with neuronal excitability.17
In humans, positron emission tomography (PET) imaging has revealed that focal seizure‐onset zones are hypometabolic during the interictal period and hypermetabolic during seizures,18 highlighting the prominent role of glycolysis in the dynamic metabolic changes seen in the epileptic brain.
Glycolysis Inhibition—A Novel Approach to Seizure Control
To mimic the physiologic effects of fasting, the KD restricts dietary carbohydrates, thereby generating ketone bodies as the proximate energy source and thereby resulting in glycolysis reduction (Fig. 1). This effect, plus the observation that minimal carbohydrate intake can abolish seizure control achieved by the KD, suggests that inhibitors of glycolysis may mimic some of the favorable therapeutic effects of the KD. That is, ketone bodies may decrease cellular excitability by decreasing glycolysis.5
The glucose analog 2‐deoxy‐d‐glucose (2DG) is a partial inhibitor of glycolysis that is a promising novel agent for seizure protection. Structurally, 2DG differs from glucose by removal of a single oxygen atom from the 2‐position (Fig. 1). 2DG is taken up by cells and undergoes phosphorylation at the 6‐position to 2DG‐6P, but glycolytic flux is reduced because 2DG‐6P cannot undergo isomerization by phosphoglucose isomerase. Uptake of glucose and 2DG is enhanced in energetically active cells.
Fluorinated 2DG has been used for several decades as a tracer (18F‐2DG) for measurement and imaging of regional glucose utilization by PET.19 2DG has also been investigated as adjuvant chemotherapy for several types of cancer, with the rationale that rapidly dividing, metabolically active neoplastic cells with enhanced glucose uptake are particularly vulnerable to glycolytic inhibition by 2DG.20, 21
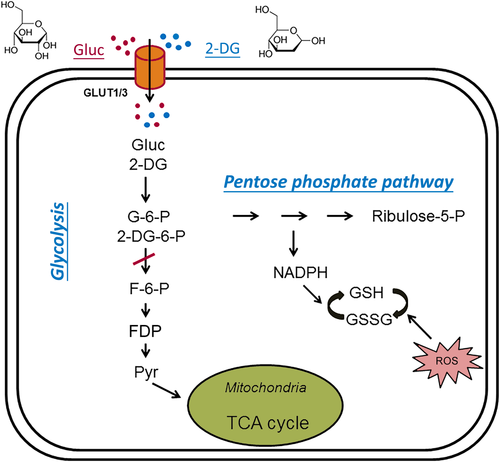
Figure 1. Glycolytic inhibition via 2‐deoxy‐d‐glucose (2‐DG) and fructose‐1,6‐diphosphate (FDP), both of which decrease glycolytic flux, reduce cell energy production, and produce antiseizure effects. Glucose (Gluc) and 2‐DG enter the cytoplasm via glucose transporters (GLUT3 in neurons or GLUT1 in glia). During glycolysis, glucose is first phosphorylated to glucose‐6‐phosphate (G‐6‐P), which is then converted into fructose‐6‐phosphate (F‐6‐P). F‐6‐P is converted into fructose‐1,6‐diphosphate (FDP) and ultimately to pyruvate via multiple steps. Pyruvate is the final product of glycolysis; it enters mitochondria to participate in the tricarboxylic acid (TCA) cycle for oxidative ATP production. G‐6‐P also enters the pentose phosphate pathway (PPP), generating reduced nicotinamide adenine dinucleotide phosphate (NADPH) and glutathione (GSH), which attenuates cell damage caused by reactive oxygen species (ROS). In addition, GSH may have antiseizure properties. Once inside the cell, 2‐DG is phosphorylated to 2‐DG‐6‐P, which cannot be further metabolized. By inhibiting phosphoglucose isomerase, 2‐DG limits the conversion of G‐6‐P to F‐6‐P, which is the main mechanism of glycolytic inhibition by 2‐DG. The chemical structures of glucose and 2‐DG are indicated at the top of the figure.
2DG Exerts Antiseizure Actions In Vitro and In Vivo
The effects of 2DG have been tested in several models of acute seizures in vivo and in vitro.22 When extracellular potassium is increased (K0+, 7.5 mM), hippocampal CA3 neurons in slices from adult rats develop high frequency interictal (epileptiform) bursting (~30/min). 2DG, when added to the bathing medium, reversibly reduced the burst frequency by about half.22 Likewise, 2DG decreased epileptiform bursts induced by bath application of the chemoconvulsants bicuculline, a γ‐aminobutyric acid (GABA) receptor antagonist, or 4‐aminopyridine (4‐AP), a potassium channel blocker.
In slices from immature animals (postnatal days 10–13 [P10–13]), 7.5 mM K0+ induced prolonged ictal discharges in the CA3 subfield for 10–30 s; 2DG decreased the ictal burst frequency. The effect of glycolytic inhibition on neuronal firing was tested in spontaneously active hippocampal neurons (CA3) when synaptic transmission was left intact or blocked with alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazoleproprionic acid (AMPA), N‐methyl‐d‐aspartate (NMDA), and GABAA receptor antagonists (6,7‐dinitroquinoxaline‐2,3‐dione (DNQX), 2‐amino‐5‐phosphonovalerate (APV), and bicuculline, respectively).23 Under both conditions (synaptic activity intact or blocked), bath application of 2DG inhibited spontaneous firing in 67% of CA3 pyramidal neurons. In contrast, CA3 neuronal firing persisted when 2DG was applied intracellularly, suggesting that glycolytic inhibition of individual neurons is not sufficient to stop neuronal firing.
The effects of 2DG on epileptiform network bursts in area CA3 were also tested in Mg2+‐free medium containing 4‐AP. Bath application of 2DG abolished these epileptiform bursts in a dose‐dependent, all‐or‐none manner.23 Taken together, these data suggest that altered glucose metabolism profoundly affects cellular and network hyperexcitability and that glycolytic inhibition by 2DG can effectively abate epileptiform activity. In summary, 2DG has an antiseizure effect on both interictal and ictal epileptiform activity in the hippocampal CA3 region.
2‐Deoxy‐d‐glucose also exerts an antiseizure effect in animal models of seizures. Kindled seizures were elicited by stimulation of the perforant path or the olfactory bulb.22, 24 In response to kindling of the perforant path, but not the olfactory bulb, 2DG‐pretreated rats displayed an increase in mean afterdischarge (AD) threshold over time. These results establish that the antiseizure effect in the kindling model is region‐specific.
Anti‐seizure effects of 2DG were also documented in 2 other acute seizure models in animals. First, in the 6‐Hz stimulation model,25 psychomotor seizures were induced by corneal stimulation using electrical pulses at a frequency of 6 Hz. Pretreatment with 2DG resulted in seizure protection in 75% of rats tested.22 Second, in Fring's audiogenic seizure‐susceptible mice, 50% of animals were protected from sound‐induced seizures after pre‐treatment with 2DG.22
However, 2DG did not protect against pentylenetetrazole (PTZ)– or maximal electroshock–induced seizures in rats, so 2DG does not exert a universal antiseizure action. 2DG's pattern of effectiveness is unlike that provided by any conventional antiseizure medication. Thus 2DG's mechanisms are likely to be broad, affecting seizure‐induced plasticity across many seizure types and syndromes, but it has not yet been possible to establish an exact correlation with human epilepsies.26
Notwithstanding the aforementioned antiseizure effects demonstrated in several investigations, some studies have not only failed to verify that 2DG exerts an antiseizure effect but have reported that 2DG actually worsens seizures. It is essential to establish whether 2DG is actually proconvulsant or whether methodologic or interpretational confounds led to that conclusion. In mice exposed to certain chemoconvulsants (PTZ, kainic acid), in the presence of 2DG, latency to seizure onset was slightly decreased.27 These findings were interpreted as a proconvulsant effect of 2DG.
However, an alternative explanation is that 2DG increases cerebral blood flow acutely,28 enhancing delivery of convulsants to the brain and thereby shortening the seizure threshold and latency. In another study, by Samokhina et al.,29 chronic reduction of glucose utilization by daily intracerebroventricular administration of 2DG for 4 weeks reportedly had a proconvulsant action. Specifically, 2 of 10 rats treated with 2DG chronically developed spontaneous seizures; the other 8 2DG‐treated rats did not develop spontaneous seizures. This is hardly a robust effect.
Moreover, in that study, the 2 rats that did develop spontaneous seizures had a huge number of seizures: 26 to 42 per hour, lasting up to a minute in duration. It is unclear why these 2 rats had so many seizures, whereas the 8 nonseizing rats had no spontaneous seizures. If 2DG is inherently proconvulsant, such a dichotomous all‐or‐none result would not be expected. Second, these authors reported that evoked excitatory synaptic potentials in chronically treated rats were reduced, which may lead to anticonvulsant rather than proconvulsant effects. Third, a technical concern is that chronic implantation of the intraventricular cannula (for 2DG injection) is an invasive approach and may itself cause brain injury that could potentially lead to the genesis of posttraumatic epilepsy in some animals.
Another study tested the effects of 2DG on rat hippocampal slices, wherein NMDA receptors and GABAA receptors were blocked, and cesium was used to block all potassium channels.30 In this experimental protocol, 2DG appeared to induce ictal‐like bursting, although interictal epileptiform activity was suppressed. Any conclusions about a potential proconvulsant effect of 2DG under such pharmacologically extreme conditions should be approached with caution.
Potential Alternative Antiseizure Actions Mechanisms of 2DG versus the KD
KD and 2DG both suppress seizure activity, but they likely work through different (although perhaps overlapping) mechanisms. It is important to note that 2DG does not cause ketosis. Although 2DG and the KD suppress seizures in Fring's mice and in the 6‐Hz model (both models of focal‐onset seizures),31 only the KD is effective in MES (a model of generalized tonic–clonic seizures).
Kindling is abrogated by both the KD and 2DG, although prior studies utilized somewhat different induction protocols.22, 24, 32 Therefore, 2DG and the KD are not comparable directly, although both modify seizure susceptibility by altering metabolic pathways involved in energy regulation. Studies with both 2DG and the KD can provide potentially useful clinical information.
2DG is Antiepileptic and Neuroprotective
Antiepileptogenesis is defined as the slowing or prevention of epilepsy development (“disease‐modification”). The anti‐epileptogenic effect of 2DG was studied using olfactory bulb or perforant path kindling. Kindling progression (as opposed to acute kindled seizures, described in previous sections) was quantified by the number of ADs required to elicit each defined stage of seizure severity, according to the Racine scale.33
Intraperitoneal pre‐treatment with 2DG at doses ranging from the minimally effective dose of 37.5 mg/kg up to 250 mg/kg, given 30 min before each kindling stimulation, resulted in a 2‐fold increase in the number of ADs required to achieve class III, IV, or V kindled seizures in both brain regions.22, 24, 34 These results verify 2DG's antiepileptic action.
Furthermore, 2DG slowed the progression of kindled seizures when given immediately after or 10 min after a kindled seizure, raising the possibility that this agent may be useful for seizure clusters or even status epilepticus.34
In other models, 2DG has also been shown to exert neuroprotective effects. For example, in hippocampal cell cultures, 2DG enhanced the resistance to oxidative and metabolic insults by inducing stress proteins and protected against kainic acid seizure‐induced hippocampal neuron loss and cognitive impairment in rats.35
In the bilateral carotid artery occlusion model in mice, 2DG pre‐treatment reduced seizure occurrence.36 Potential mechanisms of 2DG‐mediated neuroprotection include activation of adenosine monophosphate–activated protein kinase,37 reduced oxidative stress,38 and protein unfolding secondary to disruption of glycosylation.39
Potential Mechanisms of 2DG Action
The acute and chronic actions of 2DG likely entail different cellular and molecular mechanisms. Chronic antiepileptic effects of 2DG have been attributed to decreased expression of brain‐derived neurotrophic factor (BDNF) and its receptor, tyrosine kinase B (trkB), both required for kindled seizure expression.40
2DG suppresses seizure‐induced increases in BDNF and trkB, mediated by the transcriptional repressor neuron restrictive silencing factor (NRSF) and its nicotinamide adenine dinucleotide hydride (NADH)–sensitive co‐repressor carboxy‐terminal binding protein (CtBP), which acts at the promoter regions of the Bdnf and TrkB genes.24 During seizures, glycolysis increases to meet the energy demands of actively firing neurons.
The increase in glycolysis raises NADH levels, in turn causing CtBP to dissociate from NRSF, resulting in decreased transcriptional repression and increased BDNF and trkB levels. When glycolysis is reduced in the presence of 2DG, NADH levels fall, so the NRSF‐CtBP complex maintains its repression of BDNF and trkB, and kindling progression is retarded.24, 41 NRSF is not required for the antiepileptic effect of the KD, despite its crucial role in mediating chronic 2DG effects—in NRSF‐knockout mice, the anti‐epileptic effect of 2DG was abolished but the KD still afforded protection against kindling progression.42
Further evidence linking metabolism to cellular excitability comes from experiments showing that the effect of 2DG is mediated through upregulation of ATP‐sensitive potassium (KATP) channel subunits Kir6.1 and Kir6.2.43, 44 KATP channels are closed when intracellular ATP is high and open when intracellular ATP is low.
Open KATP channels efflux K+, hyperpolarizing the cell and decreasing its excitability. Decreased ATP levels, if spatially restricted to areas of the membrane subjacent to KATP channels, might lead to enhanced K+ efflux and hyperpolarization. It has been proposed that pannexin (Panx1) channels are involved in the sensing of intracellular glucose levels and extracellular ATP release; ATP then dephosphorylates to adenosine, producing hyperpolarization.45
An intriguing possibility is that the metabolic control of excitability varies by brain region or cell type, comprising “gates” to filter pathologic discharges (e.g., substantia nigra, dentate gyrus).5, 44, 46
Some data suggest that the antiseizure effects of 2DG in vitro and in vivo are quite rapid,47, 48 implying that 2DG may exert direct actions at membrane or synaptic levels, whereas others report the more gradual onset of an antiseizure effect.49 The effects of 2DG on excitatory synaptic transmission were investigated in pilot experiments using hippocampal slices.47, 48 In hippocampal CA3 neurons, there was no effect of 10 mM 2DG on the frequency or amplitude of spontaneous excitatory postsynaptic currents (sEPSCs).
However, after the induction of epileptiform bursting in this region by elevated (7.5 mM) K0+, 2DG reduced sEPSC frequency and amplitude, suggesting that the effects of 2DG are activity‐dependent—specifically, 2DG may be consumed by actively firing neurons and acts preferentially when neuronal activity is high, such as during seizure activity.
The same 2DG dose had no effect on miniature EPSCs isolated by exposure to tetrodotoxin, which blocks sodium channels and thus neuronal activity‐mediated synaptic release of neurotransmitters, but miniature EPSCs were significantly reduced by 10 mM 2DG when K0+ was elevated. Because other glycolysis inhibitors depress sEPSCs in both normal and epileptic slices, the preceding results cannot be ascribed to a simple result of glycolysis inhibition.50, 51
2DG also reduces the frequency of spontaneous inhibitory postsynaptic current (sIPSC) when K0+ is elevated (7.5 mM), but to a lesser extent than sEPSCs, resulting in an overall decrease in excitatory neurotransmission.52 2DG does not affect intrinsic membrane properties significantly, and its acute effects on synaptic transmission appear to be primarily presynaptic.23, 52 In a 4‐AP model of neocortical seizures, 2DG reduced the amplitude and duration of seizures over minutes to an hour; this slower action could be related to cell death secondary to glycolytic inhibition.49
In addition to its effects on excitatory synaptic transmission, 2DG may also affect GABAergic signaling, specifically by potentiating extrasynaptic tonic GABAergic current through activation of neurosteroidogenesis.53 However, unlike glutamate in excitatory neurotransmission, GABA does not couple inhibitory neuronal activity with glucose utilization.54 Thus, although 2DG may increase postsynaptic tonic inhibition, this action appears independent of its effect on glycolysis.
The use‐dependence of 2DG action represents a critical aspect of its function. 2DG is taken up only by neurons that are metabolically active, as occurs in neuronal circuitry involved in ongoing seizures. This use‐dependence is a distinct advantage when considering the goal of a therapeutic strategy that targets only brain areas exhibiting sustained seizure activity.
2DG as a Potential Clinical Agent
Before 2DG is approved for clinical use in humans, safety and efficacy need to be demonstrated. Preclinical studies of safety and toxicity have shown that 2DG is well tolerated in rats and dogs at doses associated with antiseizure and antiepileptic effects. The Morris water maze was used to assess the acute and chronic effects of 2DG on spatial learning and memory.55
Two protocols were employed. For acute testing, 2DG was injected 15 min before water maze trials each day of testing. For chronic testing, 2DG (250 or 500 mg/kg, i.p.) was injected daily for 14 days before water maze testing was performed. There was no difference in the latency to platform acquisition (spatial learning) or retention of platform location (probe test) with either protocol, suggesting that 2DG does not significantly impair spatial learning or memory.
Open field testing was used to assess exploratory activity and anxiety. Rats were pre‐treated (30 min before testing) i.p. with saline, 50 mg/kg 2DG, or 250 mg/kg 2DG.55 Exploratory activity (number of lines crossed in the open field arena) did not differ between the saline‐ and 50 mg/kg 2DG‐treated groups, but the rats receiving 250 mg/kg 2DG had decreased motor activity (i.e., fewer lines were crossed). The reduced motor activity at 250 mg/kg was transient and reversible; if the 250 mg/kg group was later treated with 50 mg/kg 2DG, decreased exploratory activity was not seen.
These behavioral tests show that 2DG has no permanent adverse effects on spatial learning and memory, exploratory activity, or anxiety at doses that suppress seizures and retard epilepsy progression, supporting the potential of 2DG for clinical use.
Some safety studies have raised the concern that 2DG, at high doses, is associated with cardiotoxicity. In rats or mice, oral or intravenous doses of 2DG (250–2,000 mg/kg) given over 7 days were associated with a dose‐dependent decrease in mean arterial pressure and respiratory rate and an increase in mortality.56 Detailed pathologic evaluation of cardiac tissue after chronic oral 2DG ingestion in 2 rat strains revealed vacuolization of cardiac myocytes, increased incidence of pheochromocytomas, and reduced lifespans.57
The cause of these cardiotoxic effects of 2DG was most consistent with autophagy as a mechanism. Subsequent comprehensive preclinical safety and toxicology studies have demonstrated reversible species‐specific cardiotoxicity at high 2DG doses in rats but not in dogs. Fortunately, these toxic effects were detectable by measuring plasma N‐terminal pro‐brain natriuretic peptide (Nt‐proBNP) levels, offering a way to monitor for 2DG toxicity.58
Human safety of 2DG has been established in cancer trials.20 Because tumor cells are dependent on glycolysis to support their increased metabolic requirements, reducing glycolysis with 2DG deprives these cells of their requisite cellular fuel.21 2DG inhibits breast cancer cell growth in a dose‐dependent manner and causes cell death through apoptosis.59 In combination with Adriamycin, 2DG slows the growth of some solid tumors such as osteosarcoma.60
In patients with prostate cancer, a 2‐week dose‐escalation study revealed dose‐limiting asymptomatic QTc prolongation at 60 mg/kg, but doses of 45 mg/kg were well tolerated.61 The serum half‐life of 2DG in humans has recently been established to be in the 5 to 7 h range, similar to that in several animal species including rodents and dogs.62 The half‐life is one parameter used to determine the optimal clinical dosing schedule.
Future Strategies for Glycolytic Modulation of Epilepsy
Glycolysis inhibition with compounds such as 2DG represents a novel approach to epilepsy treatment. 2DG exerts acute antiseizure effects in vitro that are independent of the method of seizure induction. 2DG induces acute antiseizure effects in vivo in several seizure models and also exhibits novel chronic antiepileptic effects against progression of seizures and the adverse consequences of seizure‐induced plasticity that are associated with alterations in neuronal gene expression.
It is important to note that 2DG has a favorable preliminary toxicity profile. Together, these observations support 2DG or similar agents as feasible for treating epilepsy in patients. Clinical trials of 2DG in human epilepsy patients are anticipated.
Another approach to metabolic modulation involves the glycolytic intermediate, fructose‐1,6‐diphosphate (FDP). FDP has been shown to exert acute antiseizure activity in several seizure models in adult rats including kainic acid, pilocarpine, and PTZ.63 In these studies, the effectiveness of FDP as an antiseizure agent exceeded that of 2DG, KD, and valproate. FDP diverts glucose flux from glycolysis into the pentose phosphate pathway (PPP) (Fig. 1).
NADPH generated in the PPP reduces glutathione, which itself has antiseizure properties. Therefore, FDP may exert an endogenous antiseizure action.64 Subsequent studies showed that FDP retards kindling progression by attenuating BDNF and trkB expression,65 similar to 2DG. Furthermore, FDP inhibits the kindling‐induced downregulation of the expression of potassium‐chloride cotransporter 2 (KCC2) and decreases the expression of sodium‐potassium‐chloride cotransporter 1 (NKCC1), suggesting that FDP might alter the switch between GABAergic excitation and inhibition.63 Recent studies suggest that FDP blocks membrane calcium channels, perhaps in addition to an intracellular metabolic effect.66
Together, these results confirm that modification of glycolysis is a potentially feasible, novel approach to decreasing neuronal excitability. Further information regarding the antiseizure and anti‐epileptic mechanisms of 2DG, FDP, and other glycolytic inhibitors will enhance our understanding of the connection between cellular excitability and metabolism.
Acknowledgments
Dr. Rho's research has been supported by the Canadian Institutes of Health Research. Dr. Stafstrom's research has been supported by The Charlie Foundation, the Mathias Koch Memorial Fund of the Community Foundation of Southern Wisconsin, and the Sandra and Malcolm Berman Foundation.
Disclosures
We confirm that we have read the journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines. Dr. Shao has no conflict of interest to disclose. Dr. Rho has served as a paid consultant for Accera Pharma, Xenon Pharmaceuticals, Danone Nutricia, and Ajinomoto USA. Dr. Stafstrom has served as a paid consultant for Mallinckrodt and has received speaker fees from Nutricia. Drs. Rho and Stafstrom have received royalties from UpToDate for a chapter about the mechanisms of seizures and epilepsy. Dr. Stafstrom is an inventor on a patent application for the use of 2DG as a clinical antiseizure medication through the Wisconsin Alumni Research Foundation.



