






September 2017
Marina Diotallevi Paola Checconi Anna Teresa Palamara Ignacio Celestino Lucia Coppo Arne Holmgren Kahina Abbas Fabienne Peyrot Manuela Mengozzi Pietro Ghezzi1
Glutathione (GSH), a major cellular antioxidant, is considered an inhibitor of the inflammatory response involving reactive oxygen species (ROS). However, evidence is largely based on experiments with exogenously added antioxidants/reducing agents or pro-oxidants.
We show that depleting macrophages of 99% of GSH does not exacerbate the inflammatory gene expression profile in the RAW264 macrophage cell line or increase expression of inflammatory cytokines in response to the toll-like receptor 4 (TLR4) agonist lipopolysaccharide (LPS); only two small patterns of LPS-induced genes were sensitive to GSH depletion.
One group, mapping to innate immunity and antiviral responses (Oas2, Oas3, Mx2, Irf7, Irf9, STAT1, il1b), required GSH for optimal induction. Consequently, GSH depletion prevented the LPS-induced activation of antiviral response and its inhibition of influenza virus infection. LPS induction of a second group of genes (Prdx1, Srxn1, Hmox1, GSH synthase, cysteine transporters), mapping to nrf2 and the oxidative stress response, was increased by GSH depletion. We conclude that the main function of endogenous GSH is not to limit inflammation but to fine-tune the innate immune response to infection.
Introduction
Several studies have concluded that oxidative stress, due to increased production of reactive oxygen species (ROS), for instance, because of infection, can trigger inflammation, although the concept has been considered an oversimplification (1).
This hypothesis is largely based on studies showing that exogenously added ROS induce inflammatory cytokines, while addition of antioxidants, including the main thiol antioxidant, glutathione (GSH), inhibits it. This led to the view of ROS as pro-inflammatory mediators and GSH as an anti-inflammatory mediator (2, 3). However, although animal studies have shown a protective effect of GSH or its precursors in animal models of inflammatory diseases, such as sepsis or acute respiratory distress syndrome (4, 5), this was not confirmed in clinical trials (6).
Furthermore, physiological concentrations of ROS, which may not result in oxidative damage, as well as changes in the redox state of cellular thiols, are implicated in biochemical signaling, and administration of antioxidants could disrupt all these redox-dependent signaling mechanisms (7–9).
While in the context of oxidative stress GSH acts as an antioxidant ROS scavenger, in the context of redox regulation the couple GSH/GSSG (oxidized GSH) acts as a signaling molecule that regulates protein function via thiol–disulfide exchange reactions including protein glutathionylation (10).
To investigate the role of endogenous GSH in inflammation, whether it acts as an antioxidant or a signaling molecule, we used the mouse macrophage RAW cell line stimulated with lipopolysaccharide (LPS) with and without pretreatment with the GSH synthesis inhibitor buthionine sulfoximine (BSO). GSH/GSSG levels were measured and LPS-stimulated ROS production was quantified by electron paramagnetic resonance (EPR). BSO is an inhibitor of GSH synthase that has been used to deplete GSH in vitro, including in macrophages (11, 12), and is more specific than GSH-depleting agents such as diethylmaleate that can activate nrf2 directly due to its electrophilic properties (13).
We analyzed the gene expression profile and identified patterns of LPS-induced genes that were inhibited by endogenous GSH or that, on the contrary, required GSH for their induction. The results indicate that, contrary to the initial hypothesis, inflammatory genes are not affected by the lack of endogenous GSH. Instead, a small pattern of genes mapping to innate immunity and antiviral activity required GSH for their induction.
Discussion
This study supports the view that endogenous GSH plays a pivotal role for the establishment of the innate immune responses to viruses, possibly acting as a signaling molecule with a mechanism different from simple scavenging of ROS. Overall, a 99% decrease in GSH levels had a minimal impact on the gene expression profile of LPS-treated macrophages; LPS regulated the expression of about 15% of the transcripts, of which only less than 4% (i.e., 0.6% of the total) were affected by BSO (Figure 2). The fact that the vast majority of transcripts were unaffected by BSO is also an indirect confirmation that, within the concentrations and incubation times used, BSO does not have significant toxic or non-specific effects.
Of the genes belonging to the category “inflammatory response” (GO:0006954), comprising several inflammatory cytokines that were induced by LPS in our model (60 transcripts at 2 h and 64 at 6 h), only one gene (CXCL10) was upregulated by BSO. The observation that GSH depletion does not exacerbate the transcription of inflammatory genes, at least in our experimental conditions, might seem at variance with the existing literature starting from pioneering paper by Schreck et al. (24), reporting that ROS activate NF-kB and increase several inflammatory genes, while thiol antioxidants inhibit their expression (25). However, most of that evidence is based on in vitro or in vivo experiments using exogenously administered thiol antioxidants or pro-oxidants. What our data do not support is the extrapolation of evidence from those experiments to the conclusion that GSH is an endogenous anti-inflammatory molecule through its ROS-scavenging activity. In fact, previous reports noted that exogenous GSH or its precursor NAC inhibits the production and expression of TNF, IL-6, and IL-8 by LPS-stimulated macrophages in the absence of any significant change in intracellular GSH (25). The results reported here are also in agreement with our previous studies where we observed that there are more H2O2-induced genes that require GSH for their upregulation than genes whose induction by H2O2 is exacerbated by GSH depletion (26). Interestingly, in that study using human monocytic cells, many of the H2O2-induced genes for which GSH had a facilitatory role were related to immunity (26).
In addition, the only LPS-induced transcripts mapping to innate immunity in their functional annotation were inhibited, rather than upregulated, by GSH depletion (Group 2 genes). Not only innate immunity genes in Group 2 require GSH for their induction but also they were not induced by ROS alone (using menadione as a ROS-generating chemical) and their LPS induction was not inhibited by NAC, ruling out the possibility that ROS act as signaling molecules in their induction by LPS. The only exception was il1b whose LPS induction was inhibited by NAC but was also inhibited by GSH depletion, suggesting that GSH is important for IL-1b induction by LPS but possibly not through an antioxidant mechanism because (i) exogenous NAC and endogenous GSH appear to have an opposite role, and (ii) an oxidant alone does not induce IL-1b expression. In line with these findings, it has been shown that molecules altering intracellular thiol content with different mechanisms (i.e., GSH vs NAC derivatives) are able to influence differently LPS-induced pathways (7).
The innate immune response is also important for antiviral defense and activation of TLR4 leads to induction of antiviral proteins including IFNs and IFN-related genes (27, 28) such as MxA and Oas (29, 30). Our data, although obtained in a model where infectivity was low, suggest that GSH is important for the activation of an antiviral response. This happens without affecting inflammatory genes, except for IL-1b whose induction was also facilitated by the presence of GSH. There is evidence for a fine-tuning of TLR signaling (31), and these data indicate that GSH may be important in directing it toward specific small patterns of genes implicated in host defense rather than toward those responsible for the inflammatory response, as outlined in Figure 10.
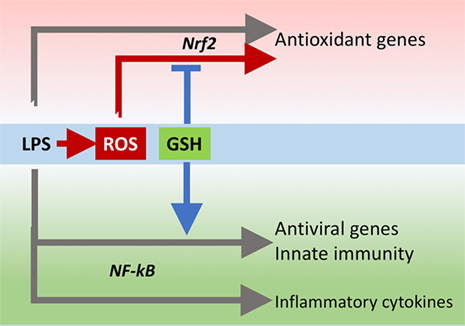
FIGURE 10. GSH fine-tuning of TLR4 signaling. LPS triggers TLR4 to induce gene expression of inflammatory cytokines, antioxidant genes, and antiviral/immunity pathways. GSH orients the TLR4-mediated changes in gene expression profile toward activation of host defense. GSH, glutathione; LPS, lipopolysaccharide; TLR4, toll-like receptor 4.
The behavior of genes in Group 1 is what one would expect. They include enzymes for GSH synthesis and antioxidant enzymes such as Prdx1, Srxn1, and Hmox. All these genes map to nrf2, a master regulator of redox homeostasis (32). Their regulation by BSO is in accordance with the hypothesis that endogenous GSH acts as an ROS scavenger because menadione induces their expression. However, NAC did not inhibit their induction by LPS, suggesting that LPS induces nrf2 target gene expression independently of the increase in ROS production. This agrees with a recent study by Cuadrado et al. showing that LPS can activate nrf2 via the small GTPase RAC1, independently of ROS (33).
In this picture, endogenous GSH might be important through other mechanisms than just scavenging ROS. In fact, nrf2 activation is dependent on oxidation of its redox sensor, keap1. While keap1 oxidation is mainly studied using ROS or various electrophiles, its thiol groups can also be oxidized by GSSG through a thiol/disulfide exchange reaction (34). It is therefore possible that the change in GSH/GSSG ratio caused by BSO (Figure 1) causes nrf2 activation by oxidation of keap1 and this adds up to the RAC1-dependent activation by LPS.
Several studies have indicated that activation of nrf2 by administration of electrophilic compounds has an anti-inflammatory effect and decreases LPS-induced transcription of other NF-kB target genes, including TNF, IL-1b, and IL-6, in RAW cells (35, 36). However, as mentioned earlier, in our experimental conditions in which nrf2 was likely activated by GSH depletion, as suggested by the increased expression of nrf2 target genes, we have not observed an effect on any inflammatory cytokine other than IL-1b. Once again, the difference might be that we did not use exogenous electrophiles to induce nrf2.
This highlights one point that is often overlooked. GSH is not just an antioxidant that participates in ROS elimination (either via its direct ROS scavenging activity or as a substrate for GSH peroxidases) but, like any other thiol including NAC, is also a reducing agent, as well as GSSG is a thiol oxidizing agent. Therefore, these two molecular species, GSH and GSSG, can regulate biological pathways in a redox-dependent manner, independently of ROS scavenging. This could happen by reversible oxidoreduction of protein thiol/disulfides, as described for keap1, but also by formation of mixed disulfides between GSH and protein cysteines. In fact, protein glutathionylation is a major mechanism of redox regulation of immunity (10, 37), affecting the function of key proteins including NF-kB (38), STAT3 (39), PKA (40), TRAF3, and TRAF6 (41), as well as participating in the release of danger signals (42, 43).
On the other hand, redox regulation often implies a role for the production of low levels of “regulatory” ROS (9). However, in this experimental model, the induction of host defense genes in Group 2 (at least those shown in Figure 7, il1b, Mx2, and Irf9) is inhibited by BSO, evidencing the need for GSH, but is not amplified by NAC, suggesting that scavenging LPS-induced ROS is not the main mechanism of action of endogenous GSH.
The finding that several genes that are important for the antiviral response, mostly part of IFN signaling pathways, including the antiviral proteins Oas and Mx2, require GSH for optimal induction by LPS adds knowledge to previous findings, indicating that GSH can inhibit viral infection (44, 45) and that viral infection causes release of glutathionylated thioredoxin and Prdx (46).
There is a large body of evidence showing the importance of GSH in immunity, including antiviral immunity (47), but so far this was ascribed to its action as ROS scavenger to inhibit oxidative stress. The present study indicates that GSH has other important signaling roles independently of protection from oxidative stress, and its action may not be vicariated by another thiol antioxidant. It might even be hypothesized that the “oxidative stress,” and consequent GSH depletion, caused by a virus as a direct consequence of its replication cycle (48) and implicated in the pathogenesis of the disease (49, 50), could be a way by which the virus attempts to diminish the antiviral response by impairing GSH-dependent antiviral pathways.
However, to understand the validity of our conclusions to other models, one needs to bear in mind the limitations of this study that is investigating mRNAs in a cell line. Future studies will need to measure the proteins of interest (for instance, IL-1b) to see whether the changes observed at the level of transcripts are reflected in changes in protein levels. To generalize the relevance of this mechanism, the observation will need to be confirmed in primary cells, including human cells, and possibly in vivo.
Author Contributions
MD, PC, MM, IC, LC, FP, and KA performed experiments. AH, PG, KA, LC, MM, FP, and AP designed and supervised experiments. MD, PG, MM, FP, and PC wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Simon Waddell for critical revision of the manuscript.
Funding
This work was supported by a fellowship program from Istituto Pasteur Italia––Fondazione Cenci Bolognetti (to PC), PRIN CUP (grant number B86516001920001 to AP), and RM Phillips Trust (to PG).



